
The year is 2021 and we are still not able to treat:
- Alzheimer’s,
- Huntington’s,
- Parkinson’s,
- and many other diseases
WHY?
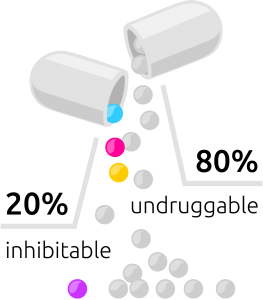
Distribution of undruggable and inhibitable drugs
The primary factor is that less than 20% percent of all pathogenic (disease-related proteins) are inhibitable. The rest is just undruggable. Ok, but what does that mean exactly?
For the sake of the argument and brevity, one should accept that a significant aspect of treating a specific disease is tackling proteins associated with that disease. Major therapeutic interventions to achieve this have come from different subdomains of biology:
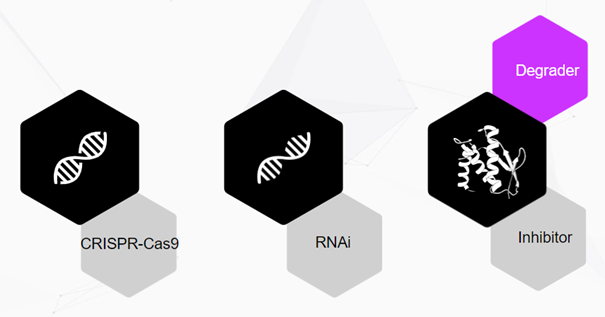
Therapeutic interventions in the past
From the genetics point of view, one can try to edit the gene associated with generating the pathogenic protein with CRISPR.
An alternative is from the ribonucleic acid (RNA) point of view. The term RNA interference (RNAi) was coined to describe a cellular mechanism that uses small pieces of RNA interacting with the genetic information-transmitting mRNA. As a result, the mRNA is cleaved into several fragments, and the information to be transferred is destroyed, or translation into a protein is prevented — a process that researchers call silencing. This, in turn, blocks potential pathogenic proteins even before their generation.
Finally, we have the inhibition approach, where a protein inhibitor is a molecule that binds tightly to a functional part of the protein, thereby inhibiting its function. Meaning the pathogenic protein gets, in effect, silenced with another compound (which can be a small molecule, peptide, or biologic drug).
As already noted above, there is only a tiny subset of pathogenic proteins that can be inhibited.
This is where targeted protein degradation comes into play, which we at Celeris Therapeutics address with AI.
How does this targeted protein degradation work from a high level, and how do we translate this to the AI domain?
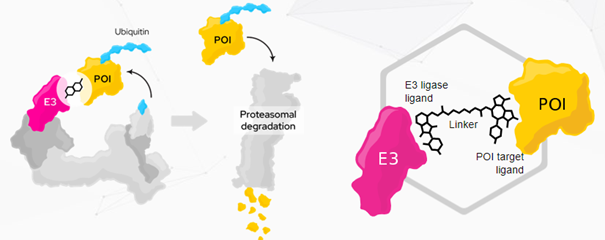
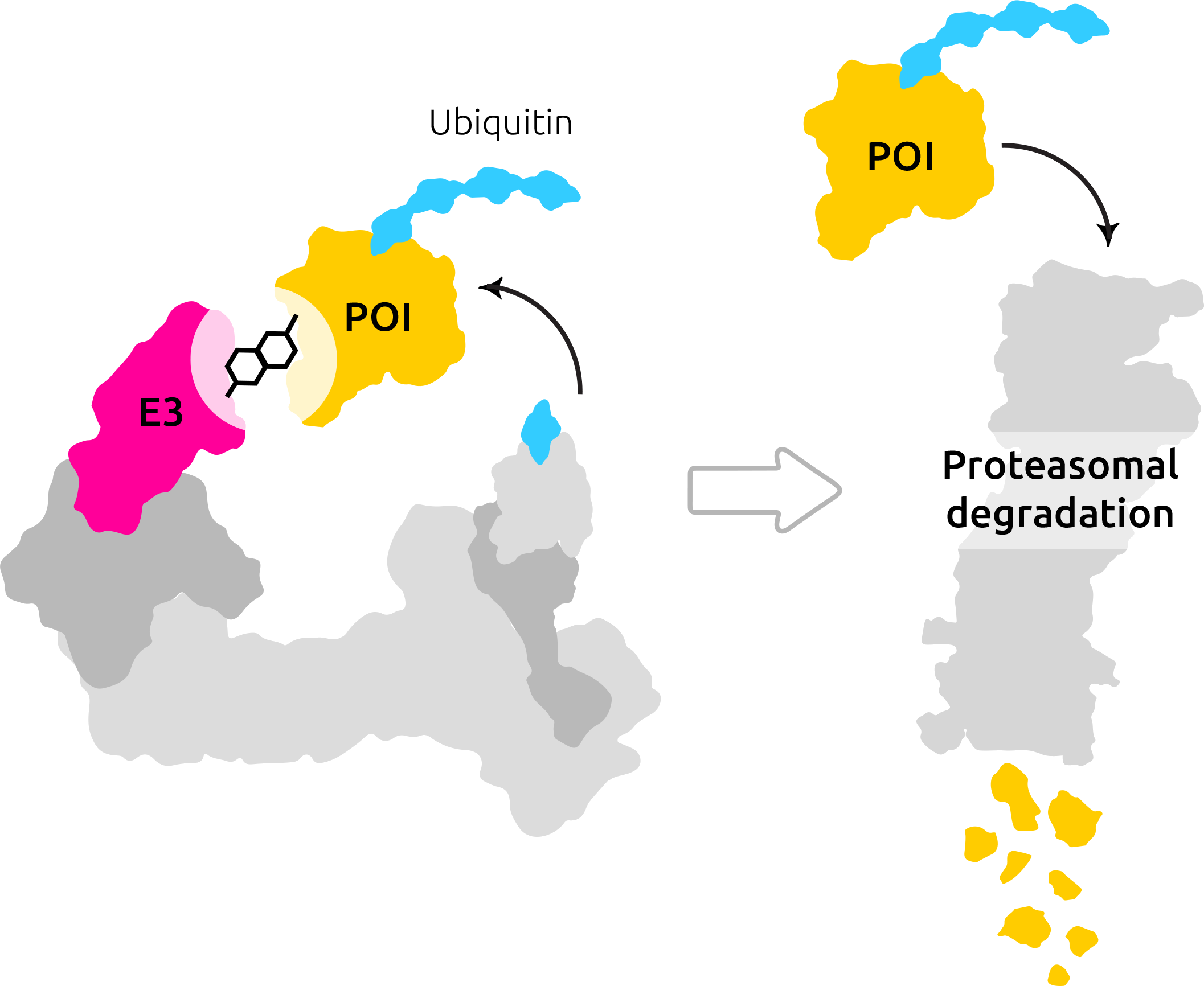
Targeted protein degradation
Degraders are heterobifunctional molecules consisting of one or two (linked) fragments (ligands) capable of binding two individual proteins simultaneously. Proteolysis-targeting chimeras (PROTAC®) are composed of two fragments and a linker connecting the two fragments.
Degrader molecules bind to an E3 ligase and a pathogenic protein (POI — protein of interest).
The binding of such a degrader molecule to a POI as well as to an E3 ligase at the same time results in the formation of a ternary complex. This ternary complex induces the targeted degradation of the pathogenic protein, as the E3 ligase triggers protein degradation via the proteasome and the ubiquitination.
Ternary complex formation in degrader function had been known for several years, as degraders that are weaker binders can also induce the degradation of proteins under the condition of ternary complex formation between a protein of interest, a degrader molecule, and a recruited E3 ligase. The significance of such ternary complexes was shown with the first ternary complex crystal structures, which displayed positive cooperativity and newly formed protein-protein interactions.
The ubiquitin-proteasome system (UPS) enables the degradation of 80% of all pathogenic proteins that are currently considered untreatable by modern drugs, as it includes the E3 ubiquitin ligase.
Short & Sweet: To tackle this problem from an AI perspective, one should understand a ternary (meaning 3 elements) complex that needs to be predicted.
- A protein of interest (Pathogenic protein)
- E3 Ligase (Special enzymatic protein for degradation of the POI)
- Molecule binding these 2, in our case PROTAC®, but it can be any molecular glue.
Furthermore, this PROTAC®molecule can be dissolved into three smaller molecules. Two ligands (fragments) and one linker, as shown in the picture above.
So, what does this mean for AI? You have essentially five molecules (some smaller, some bigger) that you need to sample from a vast molecular space (more extensive than the number of atoms in the universe btw), and you need to predict whether they are going to interact optimally. Easy right?
The challenge becomes:
- How do we make sure that all the bio-chemo-physical requirements are present in the data?
- How do I represent these molecules in a “native” way and find intelligent ML algorithms to operate on this data?
Regarding point number 1. What was essential: Interdisciplinary work
Interdisciplinary work
This means that we try to acquire the data and map bio-chemo-physical requirements directly in the data. This allows us to avoid any false assumptions that, in the end, deem our solution incorrect. Some examples of this would be:
- We are honoring the angles and conformations between these different molecular interactions in our ternary complex. Just one wrong angle, and the chemical reaction might not take place. We have to make sure that this requirement is in the data.
- We are honoring the energy between the molecules. What we are essentially trying to do is to minimize enthalpy. What does this mean, and how does this translate when we model the data?
And regarding the data there, we come to our next point. Can you predict what will the standard convolution from computer vision predict here:



Prediction?
Chances are it will group the black dog with the black cat even though a correct prediction would be the black and white cat in pictures 2 and 3.
Why is that, you may ask? In Euclidean data representation, roughly said, we model the similarity, in this case, by the number of matching pixels. Given that here, we will primarily model color and then shape, the prediction will be faulty. If we could only model more complex relationships between the objects in this picture, like hereditary, species, etc., information, we would be able to discriminate correctly.
non-Euclidean data is everywhere.
Just look at the example above, we can see that:
the shortest path between points isn’t necessarily a straight line.
Or, put in a way that is more close to machine learning,
things that are similar to each other are not necessarily close if one uses Euclidean distance as a metric (meaning the triangle inequality wont hold).
Ok but what does this mean for molecular data representation?
- Given the intrinsic properties of molecules, native way to represent them and the inter-molecular & inter-atomic relationships are graphs.
Why? Think about a molecule. While it might seem like a tiny particle, it comprises much smaller atoms with different properties. Not only that but different relationships inside a molecule and different relationships to other atoms in different molecules. So, to model these different properties and inter-molecular & inter-atomic relationships, one needs a much more complex data object. Graphs.
Why are graphs able to do this?
From a simple computer science perspective graph as a data object comprises of edges and nodes.
As such, it has been studied for decades how one can efficiently store, compute and query over graphs. Given that we are interested in every one of the atoms (nodes) and every possible complex relationship (edges), one can easily represent the molecules with their relationships with graphs. But just storing the molecular data is not enough.
What are the smart algorithms operating on these graphs?
Problem: Standard ML operations like convolution are not (usually) defined on non-Euclidean data.
Using standard machine learning algorithms (like convolutional operation) won’t work. Why? Because to “move” the convolutional operator over the tensor, one needs to define the concept of direction. Which is nonexistent in the non-Euclidian space of graphs. Hence one needs a field that studies these algorithms — graph deep learning (GDL).
The article Geometric deep learning: going beyond Euclidean data (by Michael M. Bronstein, Joan Bruna, Yann LeCun, Arthur Szlam, Pierre Vandergheynst) provides an overview of this relatively new sub-field of deep learning:
Geometric deep learning is an umbrella term for emerging techniques attempting to generalize (structured) deep neural models to non-Euclidean domains such as graphs and manifolds.

Technology enablers for AI protein degradation
Ok, we got the data, we got the intelligent algorithms from GDL, but we also took novel technology from the following disciplines:
- Data quality and quantity (for example new AlphaFold dataset shows these improvements)
- Graph deep learning (GDL)
- Active Learning (field of AI allowing for smart querying of large datasets)
- Computational power (thanks to our partners AWS & Azure)
- Better chem- and bioinformatics methods (for example, docking methods)
We are just at the beginning of our journey at Celeris Therapeutics. Other modules of our Celeris One platform take the steps after in-silico drug discovery (with computer methods) and take it to the lab (in-vitro) to be validated in animals (in-vivo) and brought to the patients. We are making a real impact.
PROTAC® is a registered trademark of Arvinas Operations, Inc., and is used under license.














